Hvordan overvåker legemiddelprodusentene produktene sine?
Definisjon Pharmacovigilance (WHO)
«The science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug-related problem.»
Tekst: Therese Myhrstad, Drug Safety Manager, Roche Norge AS
Forventningen til pasienten når hun eller han går ut av apoteket med et legemiddel i posen, er at «dette skal gjøre meg frisk»! Samtidig er pasienten vesentlig mer bevisst på bivirkninger nå enn for bare 5–10 år siden. Man er nysgjerrig, søker etter informasjon og krever svar. Risiko for at et legemiddel forårsaker uønskede, skadelige effekter kan aldri elimineres, men med kontinuerlig overvåking, evaluering og proaktiv bruk av kunnskap kan den utvilsomt minimeres.
Samler mest mulig informasjon
I møtet mellom pasient og helsepersonell kan bivirkninger med fordel gis mer oppmerksomhet enn det tradisjonelt har hatt. Ved at både pasienter og helsepersonell innrapporterer bivirkninger bidrar man til økt kunnskap om sikkerhetsprofilen til legemiddelet. Legemiddelprodusentene ønsker å samle inn så mye informasjon som mulig omkring uønskede hendelser pasienter opplever, og gjøre ny kunnskap raskt tilgjengelig for helsepersonell og pasienter.
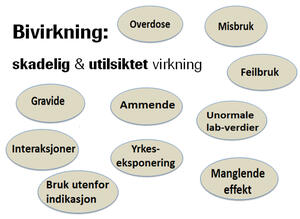
Er det kun bivirkninger som overvåkes?
Bivirkninger er den største risikoen forbundet med bruk av legemidler, men det finnes også andre potensielle risikoaspekter som inngår i overvåkingsarbeidet (figur 1) (1, 2). Ved bruk under graviditet og amming kan risiko for skadelige effekter hos henholdsvis foster og barn aldri utelukkes. Man kan via farmakologisk virkningsmekanisme og dyrestudier gjøre antakelser, men man skal alltid være årvåken overfor det uventede. Misbruk, feilbruk og overdose utgjør alle en potensiell risiko for skadelige effekter, så vel som at manglende effekt, interaksjoner og unormale labverdier også er uønskede virkninger. Farmasøyten kan søle legemiddel på huden under tilberedning, eller sykepleieren kan stikke seg på sprøyten ved et uhell, hvilket betyr at helsepersonell som eksponeres for et legemiddel i sin arbeidshverdag også kan risikere skadelige effekter.
Hvordan overvåke legemidlene?
For et legemiddelfirma er teknikkene for hvordan man overvåker nedfelt i et strengt regulert lovverk (1, 3). Lovverket er likt i EU/EØS, og overvåkingen foregår i hele produktets levetid. På laboratoriebenken tar man de første toksisitetstester, etterfulgt av dyrestudier, før man i de kliniske studiene får de første indikasjonene på hvilken sikkerhetsprofil legemiddelet har. De fleste vanlige bivirkninger identifiseres i de kliniske studiene, mens de mer sjeldne bivirkningene ofte blir kjent etter at legemiddelet er markedsført og har nådd en bredere pasientgruppe.
Bivirkningsrapportering er det viktigste verktøyet i overvåkingsarbeidet, og hele forutsetningen for at et legemiddel blir godkjent er at nytten oppveier risikoen ved å bruke legemiddelet. Denne evalueringen av «nytte-risiko» fortsetter så lenge legemiddelet er på markedet, og produsenten har også en «dynamisk» plan for videre risikohåndtering (4) som beskriver:
- Hva er identifiserte risikoer?
- Hva tror man er potensielle risikoer?
- Hva er foreløpig ukjente risikoaspekter med produktet?
Risikohåndteringsplanen angir videre de spesifikke tiltak produsenten forplikter seg til for å finne ut mer om sikkerhetsprofilen til legemiddelet.
Spontanrapporteringssystemet
Etter at legemiddelet er markedsført, innsamles mistenkte bivirkninger og de andre potensielle risikoaspektene som beskrevet i figur 1, via det som kalles «spontanrapporteringssystemet» (1, 2). Man er her avhengig av at både pasienter og helsepersonell spontant melder ifra, enten via legemiddelprodusentene eller helsemyndighetene, som videre utveksler innrapportert informasjon mellom seg.
Hva skjer med innsamlet data?
Innsamlet data danner grunnlag for kontinuerlig evaluering av nytte-risiko balansen (1, 5), som gjøres av så vel produsent som helsemyndighetene og Verdens helseorganisasjon (WHO). Resultatet av bivirkningsmeldingene kan blant annet være oppdatering av produktinformasjon, innføring av nye forsiktighetsregler eller kontraindikasjoner. Man kan finne det fordelaktig med informasjonsmateriell til pasienter eller helsepersonell utover produktomtale og pakningsvedlegg, eller se et behov for oppstart av flere studier. Indikasjoner kan innskrenkes, eller man trekker legemiddelet fra markedet.
Enkelte trender og utfordringer
Åpenhet
Økende grad av åpenhet preger legemiddelovervåkingsarbeidet. Data fra kliniske studier, bivirkningsmeldinger og sammendrag av risikohåndteringsplanene blir blant annet offentlig publisert i EU (6). De europeiske helsemyndigheter har også åpnet for offentlige høringer, hvor formålet er å få pasienters synspunkt på for eksempel akseptabel risiko versus nytte. Er denne ulik fra hvordan helsepersonell, akademia, myndigheter eller produsent tenker?
Harmonisering
Overvåkingsaktivitetene harmoniseres i økende grad på tvers av landegrensene; mer samarbeid og mer like myndighetskrav, tydeligere ansvarsområder og koordinering av informasjonsflyt.
Kunnskapstørst, egenbehandling og feilbruk
Det siste tiåret har vært preget av en stor endring i internettvanene (7). Man er konstant på nett og søker etter info, og svært ofte sjekkes helserelaterte områder. Man tar mer tak i egen helse og oppmuntres også til det. En konsekvens av dette er økt fare for egenbehandling, hvor faren for feilbruk ikke kan utelukkes. Det kan være vanskelig å navigere i mylderet av informasjon tilgjengelig på nett, og kunne skille mellom gode og mindre gode råd.
Forfalskninger
En av de største truslene for pasientsikkerheten er forfalskninger. Problemet er økende globalt, og lave strafferammer og stor fortjeneste har gjort det lukrativt for kriminelle. Er det fare for falske bivirkningsmeldinger, og dermed falsk produktinformasjon? En rekke tiltak er satt i gang for bekjempelse av forfalskninger, blant annet arbeides det for tiden med å innføre europeiske sporbarhetssystemer som kan verifisere at legemiddelet som selges på apotek er ekte (8, 9).
Underrapportering
Det estimeres at kun rundt 5 prosent av alle bivirkninger helsepersonell avdekker i sin arbeidshverdag innrapporteres (10). Nytteverdien av å melde må tydeligere synliggjøres, og må komme høyere opp på prioriteringsstigen til pasienter og helsepersonell. Er det for travle arbeidshverdager eller for tungvinte prosesser som hindrer innrapportering?
Vær en proaktiv kunnskapsrådgiver og bivirkningsmelder, bidra til kunnskapsjakten for trygg legemiddelbruk!
Referanser
- Regulation (EU) No 1235/2010.
- Guideline on good pharmacovigilance practices (GVP), module VI – Management and reporting of adverse reactions to medicinal products.
- Regulation (EU) No 536/2014.
- Guideline on good pharmacovigilance practices (GVP), module V – Risk management systems.
- Guideline on good pharmacovigilance practices (GVP), module IX – Signal management.
- Nettsiden til europeiske legemiddelmyndigheter, ema.europa.eu.
- Statistisk sentralbyrå. URL: www.ssb.no/kultur-og-fritid/statistikker/medie/_image/169305.png?_encoded=2f66666666666678302f35382f29303136286874646977656c616373&_ts=144f383e800
- Directive 2011/62/EU.
- European Federation of Pharmaceutical Industries and Associations, «European Pack Coding Guidelines».
- Norsk legemiddelhåndbok for helsepersonell. Bivirkninger og legemiddelovervåking. http://legemiddelhandboka.no/ (Sist endret: 26. september 2014).
(Publisert i NFT nr. 1/2015 side 20–21.)