Formulering av legemidler – et satsingsområde
Tekst: Hanne Hjorth Tønnesen, Solveig Kristensen og Jan Karlsen, Seksjon farmasi, Farmasøytisk institutt, Universitetet i Oslo
Innledning
Hvert år bruker legemiddelfirmaene flere milliarder kroner på å utvikle nye og mer sofistikerte virkestoff (API; active pharmaceutical ingredient) for å kunne følge med i den harde konkurransen på legemiddelmarkedet. Det er imidlertid velkjent at mange nye og tilsynelatende lovende API aldri når frem til forbrukeren. Selv et virkestoff med «perfekte» egenskaper in vitro kan bli forkastet i kliniske studier på grunn av dårlige farmakokinetiske egenskaper eller manglende målstyring in vivo. Inntil nå har det vært fokusert mye sterkere på teknologi og metoder for utvikling og modifisering av nye API enn på formulering (for eksempel valg av hjelpestoffer) og formuleringsteknologi (for eksempel legemiddelbærere). Et økt fokus på formuleringsaspektet ved utvikling av nye legemidler er imidlertid nødvendig for å øke sannsynligheten for å bringe et nytt virkestoff på markedet og for å lykkes med å lage nye generika. I denne artikkelen ønsker vi å diskutere betydningen av formulering som satsingsområde.
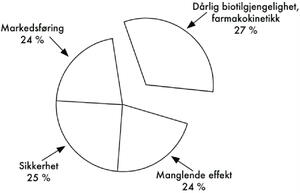
Figur 1. De viktigste årsakene til at legemidler ikke kommer på markedet (2007).
Problemstilling
Det er anslått at 38 % av legemidlene som går til klinisk utprøvning ikke passerer fase 1 (Brewster, 2007). Av dem som er tilbake elimineres 60 % i fase 2. Av dem som nå er tilbake elimineres 40 % i fase 3 og av dem som fremdeles er aktuelle blir bare 23 % godkjent av FDA (tall fra 2006). Den viktigste årsaken til at et legemiddel ikke godkjennes er dårlig biotilgjengelighet og/eller dårlige farmakokinetiske egenskaper (Wilding, 2007) (figur 1). I denne sammenhengen har man innført begrepet «drugability», dvs. en kombinasjon av basisegenskaper som legemiddelmolekylet må ha for 1) å kunne inkorporeres i en formulering og 2) å ha en forventet effekt in vivo (Brewster, 2007). «Drugability» omfatter krav til løselighet og oppløsningshastighet, fysisk form (f.eks. krystallform, partikkelstørrelse), membranpermeabilitet og stabilitet (f.eks. autoklaverbarhet) (figur 2).
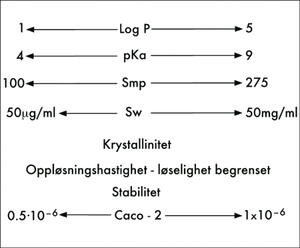
Figur 2. Eksempler på egenskaper og parametre som inngår ved vurdering av «drugability» for et nytt virkestoff.
LogP = logaritmen til vann/oktanol fordelingskoeffisienten
pKa = pH ved 50 % dissosiasjon
Smp = smeltepunkt
Sw = vannløselighet
CaCo-2 = cellelinje brukt ved permeabilitetsmålinger
De fleste legemidlene på markedet er beregnet til oral administrasjon. For orale legemidler er det utviklet et biofarmasøytisk klassifiseringssystem (BCS-systemet) som inndeler legemidlene i fire hovedgrupper basert på en kombinasjon av løselighet og membranpermabilitet (Amidon et al., 1995, Yu et al., 2002, Loftsson, 2002) (figur 3). Eksempler på API innen de ulike gruppene er vist i tabell 1 (Benet, 2007). Flertallet av dagens legemidler finnes i gruppe 1 og 2, hvorav hoveddelen er i gruppe 1 (figur 4). Det betyr at disse virkestoffene både har høy løselighet og høy membranpermeabilitet, og de kan fremstilles for eksempel i form av raskt henfallende tabletter eller tabletter med forsinket legemiddelavgivelse. Det er utvikling av disse produkttypene som mange sannsynligvis forbinder med begrepet «formulering». Dagens utfordringer når det gjelder formulering er imidlertid svært forskjellige fra hva de var for noen år siden. Dette skyldes en sterk økning av nye API i gruppe 2 og 4 (Brewster, 2007) (figur 4). Blant nye, småmolekylære legemidler under utvikling finner man bare 5 % i gruppe 1, dvs. at de har egenskaper som gjør dem relativt uproblematisk å formulere. Andelen av nye, småmolekylære legemidler i gruppe 2 er forventet å stige til 70 % og andelen i gruppe 4 til 20 % i løpet av kort tid. Felles for legemidler i gruppe 2 og 4 er at de har dårlig løselighet, og 90 % av nye API under utvikling for oral administrasjon har altså et løselighetsproblem! Tallene omfatter ikke bioterapeutika som proteiner og peptider, men man vet at desto større molekylene er, desto vanskeligere er de å formulere (Scott, 2006). Gjennomsnittlig molekylvekt for legemidler som er i fase I kliniske studier har vist en signifikant økning de siste årene selv om bioterapeutika ikke medregnes (Wenlock et al., 2003). Bioterapeutika vil i tillegg kreve alternative doseringsformer, oftest til parenteral administrasjon. Det medfører nye utfordringer i formuleringsprosessen.
| Tabell 1. Eksempler på legemidler innen de ulike BCS-gruppene | |||
| Klasse 1 | Klasse 2 | Klasse 3 | Klasse 4 |
|
Buspirone |
Atorvastatin |
Amoksicillin |
Amphotericin B |
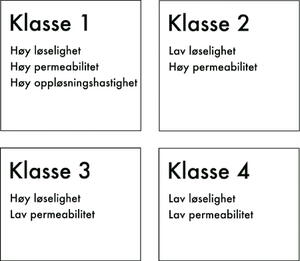
Figur 3. Klassifisering av legemidler i henhold til BCS systemet. Høy løselighet: Høyeste legemiddeldose er løselig i ≤250ml vann i pH-området 1–7. Høy permeabilitet: ≥90 % av administrert dose (i løsning)absorberes in vivo relatert til intravenøs dose eller basert på massebalanse beregninger. Høy oppløsningshastighet: ≥85 % av deklarert dose oppløses innen 30 minutter i henhold til standardisert test for oppløsningshastighet.

Figur 4. Fordeling innen BSC-gruppene for legemidler på markedet og småmolekylære legemidler under utvikling (NME).
Preformulering og estimering av «drugability»
Det er viktig å få grundig kjennskap til de fysikalsk-kjemiske egenskapene til en ny API så tidlig som mulig i utviklingsprosessen. Preformuleringsstudier utgjør anslagsvis de to første årene av utviklingsarbeidet, og omfatter en vurdering av substansens «drugability», samt de kriterier som må oppfylles for å lage en hensiktsmessig formulering (Gibson, 2004).
Følgende spørsmål bør avklares da de har stor betydning for utviklingen av produktet:
- Er det sannsynlig at biotilgjengeligeten vil være begrenset av oppløsningshastigheten eller av dårlig løselighet?
- Er det sannsynlig at løseligheten er begrenset av legemiddelets fordelingskoeffisient (logP) eller av smeltepunktet?
- Er det sannsynlig at permeabiliteten over en membran er begrensende på absorbsjonen?
Oppløsningshastigheten vil vanligvis være det hastighetsbestemmende trinnet i absorbsjonsprosessen når vannløseligheten er <100 µg/ml ved fysiologisk pH. Ved Pfizers avdeling i Groton har de undersøkt 90 000 potensielle virkestoff i løpet av 10 år og funnet at 35-40 % har en vannløselighet <10 µg/ml ved pH 7 (Lipinski, 2005). Lav oppløsningshastighet er således forventet å utgjøre et større problem enn lav løselighet for mange nye legemidler med dårlig biotilgjengelighet, og formuleringen må tilpasses etter dette.
Løseligheten av et nytt API kan estimeres ut fra følgende likning:
log Sest = - 0.01 (smp - 25) - log Pokt/vann + 0.80 (likning 1)
Her er log Sest logaritmen til den estimerte løseligheten, smp er smeltepunktet til virkestoffet og log Pokt/vann er virkestoffets fordelingskoeffisient mellom oktanol og vann.
Likning 1 viser at i tilfeller hvor smeltepunktet er høyt og fordelingskoeffisienten er lav, så vil manipulering av vehikkel (dvs. formuleringen som omgir virkestoffet) ha liten effekt på løseligheten. I slike tilfeller må egenskapene til det faste stoffet endres, for eksempel ved valg av en polymorf form med høyere løselighet eller ved fremstilling av et vannløselig salt (Myrdal og Yalkowsky, 2002). Dersom smeltepunktet er lavt og fordelingskoeffisienten høy, så vil en endring av vehikkel kunne påvirke løseligheten og eventuelt oppløsningshastigheten av API. I tilfeller hvor både smeltepunktet og fordelingskoeffisienten har høy verdi, så har man et virkestoff med dårlig «drugability» og derved en betydelig formuleringsmessig utfordring.
Membranpermeabilitetsstudier i preformuleringsfasen omfatter ofte CaCo-2-cellemodellen (Ashford, 2002).CaCo-2-celler er en human kolon karsinom cellelinje som har vært benyttet som modell for oral legemiddelabsorbsjon i snart 30 år. Ut fra diffusjonsstudier av legemiddelet over et monocellulært lag av CaCo-2-celler kan man beregne en tilsynelatende permeabilitetskoeffisient for virkestoffet. Det er observert en sammenheng mellom denne verdien og prosent absorbert in vivo for mange virkestoff (figur 5) (Ashford, 2002). Et virkestoff med tilfredsstillende «drugability» kan ha en tilsynelatende permeabilitetskoeffisient i området 0.5-1 x 10-6 cm/sek (figur 2), noe som tilsvarer 20-30 % peroral absorbsjon. Løselighetsdata kombinert med CaCo-2-permeabilitetsstudier gir også indirekte informasjon om den viktigste eliminasjonsveien for legemiddelet in vivo. Nyere studier har vist at legemidler i BCS-gruppe 1 og 2 hovedsakelig elimineres via metabolisme, mens virkestoff i gruppe 3 og 4 metaboliseres i liten grad. Videre vil legemidler i gruppe 1 påvirkes lite av aktive transportmekanismer in vivo i motsetning til legemidler i gruppe 2-4 (Benet, 2007). Denne type informasjon er viktig for valg av doseringsform- og sammensetning i det videre formuleringsarbeidet.
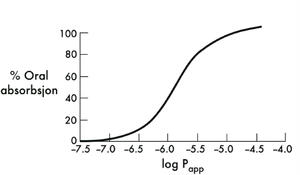
Figur 5. Sammenheng mellom logaritmen til den tilsynelatende permeabilitetskoeffisienten (Papp) beregnet i en CaCo-2-modell og prosent oral absorbsjon av et virkestoff.
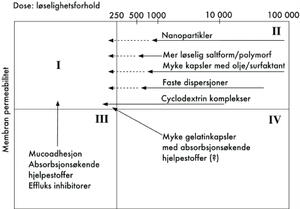
Figur 6. Eksempel på formuleringstiltak for å øke løselighet/ oppløsningshastighet og eventuelt endre løselighetsgruppe.
Formulering av API med dårlig «drugability»
Dårlige løselighets- og oppløsningshastighetsegenskaper kan forbedres ved for eksempel valg av hjelpestoffer og bruk av avansert formuleringsteknologi, mens en forbedring av membranpermeabilitet kan kreve en endring av fysiologiske forhold på absorbsjonsstedet. De største formuleringsmessige gevinstene vil derfor oppnås for API i gruppe 2 og 4. Eksempel på formuleringstiltak som kan resultere i en endring av løselighetsgruppe er vist i figur 6. En Noyes-Whitney-basert strategi representerer den vanligste løsningen når det gjelder forbedring av oral biotilgjengelighet forårsaket av lav oppløsningshastighet. Noyes-Whitney's likning beskriver oppløsningshastigheten for sfæriske partikler hvor oppløsningsprosessen er diffusjonskontrollert:
dC/dt = D * A (Cs - C) / h (likning 2)
Her er dC/dt oppløsningshastigheten, D er diffusjonskoeffisienten i det aktuelle mediet, A er det effektive overflatearealet av partikkelen som er i kontakt med oppløsningsmediet, Cs er metningskonsentrasjonen i diffusjonslaget, C er den aktuelle konsentrasjonen i mediet og h er diffusjonslagets tykkelse.
Likningen viser at oppløsningshastigheten øker dersom man øker A eller Cs. En «klassisk» Noyes-Whitney-basert strategi vil for eksempel være å øke A ved å redusere partikkelstørrelsen (mikronisering), øke Cs ved å øke stoffets vannløselighet (danne salt) og bedrepartiklenes fukteegenskaper ved tilsetning av overflateaktive hjelpestoffer.
En mer «innovativ» strategi kan være å redusere partikkelstørrelsen ved fremstilling av nanopartikler, endre partikkelstørrelse, fukteegenskaper og metningskonsentrasjon ved bruk av faste dispersjoner eller å øke metningskonsentrasjonen og eventuelt lage overmettede løsninger ved fremstilling av komplekser, co-krystaller eller bruk av amorf krystallform av virkestoffet (Peterson et al., 2006, Chokshi et al., 2007). Mikronisering av råvarer oppnås ved ulike, mekaniske knusemetoder eller ved superkritisk væsketeknologi, mens fremstilling av nanopartikler krever spesiell teknologi som forhindrer aggregatdannelse etter findeling.
Eksempler på metoder til fremstilling av nanopartikler er partikkelstørrelsesreduksjon ved kontrollert utfelling, omdannelse av krystallinsk råvare til en amorf nanodispersjon via utfelling i nærvær av en stabilisator (eks. gelatin, casein) eller ved fremstilling av polymere mikrofibre.
Eksempler på orale formuleringer hvor virkestoffet foreligger som nanokrystaller er Rapamune® (sirolimus, immunosuppressivt, 2000), Emend® (aprepitant, antiemetikum, 2003) og Megace® (megestrol acetate, progesteron, 2004) (Peterson et al., 2006, Brewster, 2007). I tilfellet med Emend® er vannløsligheten av virkestoffet ≈ 0.3 µg/ml mens daglig dose er i størrelsesorden flere hundre milligram. Her ble det tidlig vist at biotilgjengeligheten var en direkte funksjon av partikkelstørrelsen, og fremstilling av nanokrystaller gjorde det mulig å lage en tilfredsstillende formulering (Peterson et al., 2006). Nanokrystaller kan også benyttes i parenterale produkter som skal administreres intravenøst. Et virkestoff som er aktuelt i denne sammenhengen er Itrakonazol (antimycotikum). Med en vannløselighet på ~ 1 ng/ml ved pH 7 for utgangsstoffet er dette et godt eksempel på den utfordring man står overfor når kravet er å fremstille et vandig, parenteralt produkt.
En oral kapselformulering av Itrakonazol foreligger allerede på markedet (Sporanox®). Denne formuleringen består av en fast dispersjon av virkestoffet (40 %) i hydroxypropylmetylcellulose (HPMC, 60 %). Dispersjonen fremstilles ved at en løsning av virkestoffet i et organisk løsningsmiddel sprayes på små, inerte sukkerkuler (nonpareil). Kulene løses hurtig i vann, og det dannes en overmettet løsning av virkestoffet. HPMC hindrer krystallisering, og løsningen opprettholdes tilstrekkelig lenge til at legemiddelet rekker å absorberes. Så til tross for en svært lav vannløselighet, blir over 85 % av dosen absorbert fra GI-traktus og biotilgjengeligheten er >50 % (Brewster, 2007).
Som et alternativ til Noyes-Whitney basert strategi kan virkestoffet løses før administrasjon til pasient, for eksempel i en olje som deretter fylles i en myk gelatinkapsel. Legemiddelet frigjøres i GI-traktus ved hjelp av lipaser, ved solubilisering via gallesalter og ved in situ emulsjonsdannelse. Dette forutsetter at virkestoffet er løselig i et lipofilt medium, noe som ikke alltid er tilfellet. Noen virkestoffer slik som porfyrinbaserte fotosensibiliserende legemidler brukt i fotodynamisk terapi, er dårlig løselig i alle vanlige løsningsmidler uavhengig av polaritet. Typisk for disse forbindelsene er at de består av flere kondenserte benzenringer samtidig som de er switterioner. En mulighet kan da være å benytte selvemulgerende systemer (S(M)EDDS; self(micro)emulsifying drug delivery systems) dersom hensikten er å lage et oralt preparat. I tilfeller hvor «uløselige» stoffer skal fremstilles for parenteral administrasjon kan det være nødvendig å benytte en kombinasjon av flere formuleringsprinsipp. Et eksempel er parenteral emulsjon med Amphotericin B hvor man starter med å lage en suspensjon av virkestoffet for så å ende opp med en emulsjon etter flere formuleringstrinn (Desai, 1996).
Formulering av API med god «drugability»
Virkestoff som i utgangspunktet har god «drugability» kan også by på formuleringsmessige utfordringer. Årsaken kan være at det stilles spesielle krav til produktet slik som målstyring av virkestoffet til et spesifikt vev eller organ, eller krav om jevn frisetting av virkestoff over en lang periode. Hormonpreparater er et godt eksempel på dette. De vanligste steroidene slik som estradiol og derivater av dette, er virkestoff som kan klassifiseres i BCS-gruppe 1 (unntaksvis gruppe 2).
Konvensjonelle tabletter er basert på en triturasjon av virkestoffet i laktose siden API er høypotent og foreligger i svært liten mengde per tablett. Laktosen bidrar også til et hydrofilt miljø slik at virkestoffet løses raskt (Wen og Qiu, 2006). Tabletter som eneste formuleringsprinsipp er imidlertid ikke tilstrekkelig for å møte forbrukerenes krav og sikre maksimal compliance. Det finnes derfor en rekke avanserte formuleringer med hormoner på markedet, for eksempel transdemalt plaster som avgir konstant dose over et gitt antall dager (Evorel®, Estraderm®, Climara®), intramuskulær depotinjeksjon som avgir virkestoff i løpet av tre måneder (Depo-Provera®), implantat som avgir levonorgestrel 17 µg/dag over en periode på fire år (Norplant-II®), polymerbasert vaginalinnlegg som avgir konstant dose over et gitt antall uker eller måneder (NuvaRing®, Estring®), spiral hvor legemiddelet foreligger som en mikrokrystallinsk suspensjon i flytende silikon innkorporert i en hastighetskontrollerende membran og frigjøres i løpet av et år (Progestasert®) og vaginalgel basert på en oljeløsning av virkestoffet som er emulgert inn i gelen (Crinone®) (Felleskatalogen, 2007, Florence & Attwood, 1998, Chien, 1992).
Disse eksemplene understreker behovet for avansert formulering/formuleringsteknologi selv for API med gode løselig- og oppløsningshastighetsegenskaper.
Formulering av bioterapeutika
I løpet av de siste 15 årene har det vært en sterk økning i antall registrerte produkter basert på bioteknologisk fremstilte virkestoff. FDA godkjente tre bioterapeutika i 1992 og 35 slike produkter i 2002 (Baron and Massey, 2004). Bioterapeutika (for eksempel antistoffer, hormoner, DNA, gener) er svært komplekse molekyler med molekylvekt opp til 1000 ganger et vanlig, kjemisk fremstilt API (Jacques, 2006).
Virkestoffene fremstilles vanligvis fra cellekulturer og prosessen involverer grundig opprensning før et API med akseptabel kvalitet foreligger. Den komplekse prosedyren resulterer i API med en pris per gram som er 20-200 ganger høyere enn et gjennomsnittlig, kjemisk fremstilt virkestoff (Jacques, 2006). Det økonomiske aspektet, samt begrenset tilgang på virkestoffet resulterer i redusert mulighet for å «prøve og feile» i formuleringsarbeidet. Samtidig setter legemidler med protein og nukleinsyrekarakter store krav til formuleringen.
Stabilitetenav virkestoffene er ofte meget kort, og man er i tillegg avhengig av målstyrte formuleringer for å kunne utvikle farmasøytisk og medisinsk akseptable preparater. Det er helt nødvendig at formuleringsprosessen er forankret i grunnforskning for å oppnå produkter med nødvendig stabilitet, tredimensjonal-strukturell integritet og tilstrekkelig frigjøring av API på virkestedet (Scott, 2006).
De fleste bioterapeutika er fremstilt som parenterale løsninger. Hovedproblemene er løselighet (minimum 0.1-5.0mg/ml er ønskelig) og stabilitet (Scott, 2006). Produktene må være sterile og bør være isotone. Hjelpestoffer og fremstillingsmetoder som rutinemessig anvendes ved fremstilling av vanlige parenterale produkter medfører svært ofte stabilitetsproblemer i forbindelse med produksjon av bioterapeutika, og alternative metoder må utvikles. Et eksempel er nukleinsyrer (gener) som ofte formuleres sammen med gen-overføringsvektorer slik som virus, lipider eller polymere. Virale vektorer kan betraktes som store proteiner i formuleringssammenheng, og stabilisering av genmaterialet omfatter derfor også stabilisering av overføringsvektoren. Faktorer som agglomerering, adhesjon, følsomhet overfor rysting, temperatur, ionestyrke og spormetaller blir av stor betydning (Scott, 2006).
Hvert molekyl er unikt, og det er vanskelig å trekke analogslutninger vedrørende valg av hjelpestoff og fremstillingsmetode. Lyofilisering ved fremstilling av et pulver som kan løses i sterilt vann umiddelbart før bruk, kan øke stabiliteten av produktet. Det er imidlertid ikke uproblematisk å fjerne vannet ved frysetørking, og mange biomolekyler mister tredimensjonal-strukturell integritet (og derved biologisk effekt) i prosessen. Alternative administrasjonveier som transdermal, transmucosal og pulmonar tilførsel er under kontinuerlig vurdering, men medfører ytterligere formuleringsrelaterte utfordringer.
Målstyring av legemidler
Formulering omfatter også utvikling av målstyrte produkter som frigjør legemiddelet spesifikt på virkestedet. Dette kan oppnås ved å inkorporere virkestoffet i en «bærer» (f.eks. liposom, mikropartikkel, cyclodextrin, viral vektor) med spesielle overflateegenskaper eller egnet størrelse som befordrer selektiv anrikning i vev, organer eller membraner.
Det er imidlertid ikke tilstrekkelig at virkestoffet fraktes til virkestedet; det må også frigjøres fra «bæreren» i løpet av begrenset tid og i terapeutisk dose. En mulighet er å benytte «bærere» som reagerer på ytre eller indre stimuli, slik som temperatur, lys, pH eller enzymer (Baldursdottir et al., 2002). Valg av hjelpestoff i kombinasjon med type teknologi ved fremstilling av «bærer» er helt avgjørende for innkapslingsgraden av API, målsøkende egenskaper og aktiverbarhet av «bærer», samt stabilitet og mulighet for oppskalering av produktet. Utvikling av målstyrte legemidler krever evne til forskningsbasert nytenkning.
Fremstilling av nye «bærere» basert på kjente og vel karakteriserte hjelpestoff er én mulighet, slik som polymere av cyklodextriner (Ferruti, 2007). En annen mulighet er å anvende etablert teknologi på en ny måte. Fotokjemisk internalisering (PCI) er et eksempel på dette. Prinsippet for fotodynamisk terapi (PDT) som anvendes i kreftbehandling, benyttes innen PCI til å frigjøre bioterapeutika som er tatt opp i celler og oppkonsentrert i intracellulære endosomer oglysosomer (Høgset et al., 2004). For å styrke kompetansen innen området «fotoreaktivitet av legemidler» (herunder formulering, stabilitet og målstyring) er det nylig etablert et internasjonalt forskernettverk (PhoRePharm) på initiativ av avdeling for farmasi, Farmasøytisk institutt, UiO.
Reformulering og utvikling av generika
Generika er kostnadseffektive substitutter for originalpreparater. Salget av reseptpliktige legemidler basert på reformulering utgjør ca. fem ganger mer av omsetningen enn nye legemidler i USA (Thornton, 2007). Generika utgjør ca. 50 % av det amerikanske legemiddelmarkedet (Bansal et al., 2006). Kostnadene ved utvikling av et nytt originalpreparat ble estimert til 802 millioner US$ i 2006 med en gjennomsnittlig utviklingstid på 14 år, mens tilsvarende tall for generika var 1-2 millioner US$ og tre-fem år (Bansal et al., 2006).
Utviklingsfasene ved fremstilling av generika består av preformulering, oppskalering, testing av bioekvivalens og søknad om registrering. Generika skal være farmasøytisk ekvivalente med originalpreparatet, dvs. at virkestoff, styrke, doseringsform og administrasjonsvei skal være identisk. Videre skal produktene være bioekvivalente, noe som betyr at de skal ha samme oppførsel in vivo.
For å tilfredsstille disse kravene vil generika formuleres slik at de har flest mulig likhetstrekk med originalpreparatet. Karakterisering av virkestoffets fysikalsk-kjemiske egenskaper, identifikasjon av «kritiske» hjelpestoff og optimalisering av fremstillingsmetoden blir av stor betydning. Dette er spesielt viktig for virkestoff i BCS-gruppe 2 og 4 hvor små endringer i valg av hjelpestoff, krystallmodifikasjon av virkestoff og fremstillingprinsipp kan påvirke løseligheten og derved biotilgjengeligheten av produktet.
Blant de 100 bestselgende generika på det amerikanske markedet i 2006 inneholdt ≈ 54 % et virkestoff med løselighetsproblemer (Bansal et al., 2006). Selv minimale endringer i mengde og type hjelpestoffer kan resultere i at kravet om bioekvivalens ikke oppfylles (Bansal et al., 2006). Det kan også være en utfordring å reformulere produktet, f.eks. ved anvendelse av ulike polymorfe former av et API, på en slik måte at bioekvivalens opprettholdes samtidig som formuleringen danner grunnlag for patenterbare forskjeller (Peterson et al., 2006). Reformulering og utvikling av bioekvivalente produkter krever derfor grundig kjennskap til alle aspekter ved legemiddelformulering.
Legemiddelformulering som satsingsområde
I de senere årene har det blitt satt et internasjonalt fokus på betydningen av formulering ved utviklingen av nye legemidler.
Basert på fakta om at de fleste nye virkestoff har løselighetsproblemer, at molekylstørrelsen på nye API øker og at den viktigste årsaken til at legemidlene ikke kommer på markedet er dårlige biofarmasøytiske egenskaper, begynner man å ta konsekvensen av at det ikke er tilstrekkelig å komme frem til et «ideelt» virkestoff ved kjemisk syntese, screening av organismer fra hav eller land, eller ved fremstilling fra cellekulturer for å oppnå et effektivt produkt.
I 2005 gikk 11 av de største universitetene i USA sammen og dannet National Institute for Pharmaceutical Technology and Education (NIPTE) (Basu, 2006). Hensikten var å fremme forskning innen legemiddelformulering, teknologi og utvikling. Samme år mottok YorkPharma i England 1 million euro fra det tyske helseministeriet for åutvikle en nanopartikulær krem for behandling av akne (Editorial, 2006).
I England har farmasøytisk institutt ved universitetet i London startet eget firma (Pharmovation, 2005) med det formål å overføre ideer og teknologi generert ved instituttet til industrielle produkter, og universitetet i Bradford har etablert Institute of Pharmaceutical Innovation (IPI) som et senter for legemiddelutvikling med hovedvekt på biofarmasøytiske formuleringsproblemer.
I Finland har man nylig etablert et senter for formulering og legemiddelutvikling (Drug Research and Development Centre) med utgangspunkt i det farmasøytiske miljøet ved universitetet i Kuopio, og i Sverige er det etablert et firma som har spesialisert seg på reformulering og optimalisering av produkter (Orexo).
Dette er bare noen eksempler på hva som skjer internasjonalt. I Norge har vi unik tilgang på nye råstoffer fra havet av medisinsk interesse, og det satses store pengesummer for å bygge opp kompetanse innen bioprospektering (Rapp, 2007). Vi har imidlertid ikke tilsvarende mulighet i Norge til å videreutvikle potensielle virkestoff og føre dem fra laboratoriet til pasient. Norges forskningsråd har fastslått at det er et presserende behov for satsing på forskning innen formulering av legemidler (Pharmaceutics) i Norge, og anbefaler at det opprettes et nasjonalt forskningsprogram innen fagfeltet (Norges forskningsråd, 2006). Det er derfor å håpe at det finnes evne og vilje til å gjennomføre disse anbefalingene, slik at det potensialet man har for eksempel gjennom bioprospektering i nord, kan videreutvikles ved bruk av nasjonale ressurser.
Referanser
Amidon, G.L., Lennerås, H., Shah, V.P., Crison, J.R. A theoretical basis for biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability.
Pharm.Res., 12, 413-420 (1995)
Bansal, A.K., Mulla, M., Kakumanu, V.K. Criticality of functional excipients and decoding methods during generic product development.
Pharm. Technol. Eur., 18(6), 34-38 (2006)
Baldursdottir, S.G., Karlsen, J., Tønnesen, H.H. Systems for pulsed and programmed drug release. Norsk Farmaceutisk Tidsskrift, 110(2), 30-38 (2002)
Baron, L.M., Massey, A. (eds) Bio-editors and reporters guide 2003-2004. Bio, Biotechnology Industry Organization, Washington, 2004
Basu, P.K. US Universities unite. Eur. Pharm. Rev., 11(1), 18-19 (2006)
Benet, L.Z. Using a biopharmaceutics drug disposition classification system as a roadmap for maximizing drug exposure. M3: Molecules, Materials and Medicines, May 20-23, 2007, Reykjavik, Iceland
Brewster, M. Physical form and formulation needs in contemporary drug pipelines. M3: Molecules, Materials and Medicines, May 20-23, 2007, Reykjavik, Iceland
Chien, Y.W. Novel drug delivery systems. Marcel Dekker, New York, 1992
Chokshi, R.J., et al. Improving the dissolution rate of poorly water soluble drugs by solid dispersion and solid solution - pros and cons.
Drug Delivery, 14, 33-45 (2007)
Desai, N. Process design methods for disperse systems; preparation and principles. Advances in the formulation and process design of injectable dispersed pharmaceuticals. Arden House Europe Conference, Cambridge, 1996
Editorial, 1 million euro grant for acne cure. Pharm.Technol.Eur., 18(9), 16 (2006)
www.felleskatalogen.no (2007)
Ferruti, P. Recent results on biocompatible and bioeliminable hydrophilic polymers. APGI-Innovation in Drug Delivery, Napoli, September 2007
Florence, A.T., Attwood, D. Physicochemical principles ofpharmacy.
Macmillan Press, London, 1998
Høgset, A., et al. Photochemical internalization in drug and gene delivery. Adv. Drug Del. Rev., 56, 95-115 (2004)
Jacques, A. Managing a biopharmaceutical supply chain. New applications for classical techniques. BioProcess Int., 4(2), 18-22 (2006)
Lipinski, C. Drug solubility: an issue here to stay? Capsugel Expert Meeting, Nashville, November 2005
Loftsson, T. Cyclodextrins and the biopharmaceutics classification system of drugs. J.Incl.Phenom.Macrocycl.Chem., 44, 63-67 (2002)
Norges Forskningsråd Pharmaceutical Research in Norway - An Evaluation, Oslo, December 2006 www.forskningsradet.no/publikasjoner
Peterson, M.L., Hickey, M-B., Zaworotko, M.J., Ø.Almarsson Expanding the scope of crystal form evaluation in pharmaceutical science. J.Pharm.Pharmaceut.Sci., 9, 317-326 (2006)
Rapp, O.M. Kalde kryp kan knekke kreften. Aftenposten torsdag 4.oktober 2007, s.15
Scott, C. Formulation Development. BioProcess International, 4 (2), 42-57 (2006)
Thornton, P. More bites of the apple. Scrip Supplement October 2007, s.21
Wen, H., Qiu, Y. Adsorption of small drug particles at the surface of large excipients. Pharm. Technol. Eur., 18 (1), 39-44, 2006
Wenlock, M.C., et al. A comparison of physicochemical property profiles of development and marketed oral drugs. J.Med.Chem., 46, 1250-1256 (2003)
Wilding, I. Approaches to a rapid transition of drug substances and formulations into human studies. M3: Molecules, Materials and Medicines, May 20-23, 2007, Reykjavik, Iceland
Yu, L.X., et al. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm.Res., 19, 921-925 (2002)
(Publisert i NFT nr. 1/2008 side 28–32.)