Registrering av legemidler prosedyrer og dokumentasjonskrav
Tekst: Sissel Woxholt
Seksjonssjef på Registreringsseksjonen, Statens legemiddelverk
Legemiddelverkets ambisjon er å være det nasjonale kompetansesenteret på legemiddelområdet, samt å bruke vår kunnskap og kompetanse om legemidler til det beste for folke- og dyrehelsen.
Aktivitetene knyttet til legemiddelgodkjenning er finansiert over statsbudsjettet. Det kreves inn ulike avgifter til statskassen, blant annet registreringsavgiften, som i størrelsesorden er ment å samsvare med statens kostnader til saksbehandling. Registreringsavgiften betales for å søke, ikke for godkjenning av søknaden.
Norge og EU
I 1994 fikk Norge en observatørrolle på legemiddelområdet i EU gjennom EØS-avtalen. Dette innebar en harmonisering av det norske regelverket og regelverket i EU. Som en følge av dette ble behovsparagrafen opphevet, og følgelig også pris som et godkjenningskriterum. Kravet om at det for nye legemidler skulle dokumenteres en bedre effekt enn eksisterende behandling falt også bort, samtidig som parallellimport ble innført.
I år 2000 ble EØS-avtalen utvidet, og representanter fra Legemiddelverket ble fullverdige deltakere av komiteer og arbeidsgrupper i EU. Gjennom utvidelsen av avtalen ble godkjenningsprosedyrene sentral prosedyre og gjensidig anerkjennelsesprosedyre tatt inn i det norske regelverket, og Norge fikk utrederstatus i disse prosedyrene på lik linje med medlemslandene.
I 2005 ble EU-regelverket revidert og en ny prosedyre, desentralisert prosedyre innført. Dette regelverket er ennå ikke implementert i Norge, men forventes implementert i løpet av våren 2009.
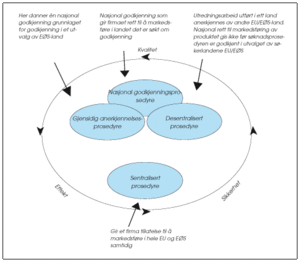
Figur 1. Kriterier for godkjenning av et legemiddel
Beskrivelse av de ulike EU-prosedyrene
Gjensidig anerkjennelsesprosedyre (MRP - mutual recognition procedure)
Utredningsarbeid utført i ett land anerkjennes av andre EU/EØS-land. Markedsføringstillatelse utstedes først som nasjonal MT i et land. Legemiddelmyndighetene i de øvrige land det søkes i, anerkjenner utredningen som er gjort for den første nasjonale markedsføringstillatelsen.
Desentralisert prosedyre (DCP - decentralised procedure)
Utredningsarbeid utført i ett land anerkjennes av andre EU/EØS-land samtidig som utrederlandet utsteder sin MT. Til forskjell fra gjensidig anerkjennelsesprosedyre sendes søknaden om markedsføringstillatelse samtidig til legemiddelmyndighetene i de landene det ønskes en markedsføringstillatelse for.
Sentral prosedyre (CP - centralised procedure)
Søknaden sendes til EMEA, the European Medicines Agency, i London som utnevner to utrederland i EØS (rapportør og korapportør) som utreder på vegne av fellesskapet. EU-kommisjonen fatter vedtak som gir søker tillatelse til å markedsføre i hele EU samtidig. Legemiddelverket er gjennom EØS-avtalen forpliktet til å fatte likelydende vedtak 30 dager etter at Kommisjonen har utstedt markedsføringstillatelsen i EU.
Følgende preparater må søkes via sentral prosedyre:
- legemidler fremstilt ved rekombinant DNA-teknologi og genmodifisering
- hybridomer og monoklonale antistoffer
- indikasjoner innenimmunsvikt, kreft, nevrodegenerativ sykdom, diabetes
- «orphan legemidler», dvs. legemidler mot svært sjeldne sykdommer
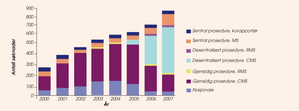
Figur 2. Antall mottatte søknader om markedsføringstillatelse per år – alle prosedyrer
Nasjonal prosedyre
Det kan søkes i nasjonal prosedyre når legemiddelet kun skal markedsføres i ett land. Kan senere utvides til flere EØS-land gjennom gjensidig anerkjennelsesprosedyre.
I de ulike godkjenningsprosedyrene vurderes dokumentasjonen med hensyn til kvalitet, sikkerhet og effekt for søknader om nye markedsføringstillatelser, endringer i markedsføringstillatelsen og fornyelser. Det har vært en stor økning i antall søknader etter at Legemiddelverket gikk inn i EØS-samarbeidet i 2000 som figur 2 viser.
(Publisert i NFT, nr. 1/2009 side 14–15.)