Bioekvivalens og misforståelser
Bioekvivalens
I artikkelen om bioekvivalens og medisinsk likeverdighet i NFT nr. 2/2009 (1) antydes det at begrepet bioekvivalens ikke forstås korrekt. Artikkelen bidrar dessverre ikke til en klargjøring av hva bioekvivalens faktisk innebærer og hvilke konklusjoner man kan trekke fra en bioekvivalensstudie, påpeker professor Eva Skovlund. Hun presiserer her hva bioekvivalens betyr, hvordan det analyseres og hvordan det skal fortolkes.
Tekst: Eva Skovlund, professor II, Avdeling for farmasøytisk biovitenskap, Farmasøytisk institutt, Universitetet i Oslo
Hva er et generisk legemiddel?
Et generisk legemiddel er et legemiddel som har samme mengde virkestoff og samme eller liknende legemiddelform som et referanselegemiddel. Godkjenningen av et generisk legemiddel baseres normalt på at bioekvivalens er vist. Bioekvivalens med referanselegemidlet må påvises med relevante biotilgjengelighetsstudier, og er med andre ord basert på en sammenlikning av farmakokinetiske egenskaper.
Bioekvivalens
Bioekvivalens kan defineres som følger: To legemidler er bioekvivalente hvis de er farmasøytisk ekvivalente eller farmasøytiske alternativer og hvis deres biotilgjengelighet etter administrasjon i samme molare dose er så lik at effekt og sikkerhet i det store og hele anses å være den samme.
De to viktigste variablene man sammenlikner for å vise bioekvivalens er areal under plasmakonsentrasjonskurven (AUC) og maksimal plasmakonsentrasjon (Cmax). I tillegg vurderes tid til maksimal plasmakonsentrasjon (Tmax) dersom dette anses for å ha klinisk betydning. Sammenlikningen er basert på forholdet (ratioen) mellom gjennomsnittlig AUC for generikum (testprodukt) og referanseproduktet (AUCtest/AUCreferanse) og tilsvarende for Cmax.
Vi må huske at man ikke kan vise at to produkter er identiske, kun at de er biologisk ekvivalente. Da er det viktig å definere hvor store avvik som skal kunne aksepteres. Som hovedregel skal et 90 % konfidensintervall for gjennomsnittlig ratio mellom test og referanse for henholdsvis AUC og Cmax være innenfor ±20 %. Fordi man beregner et geometrisk gjennomsnitt svarer dette til at man må sikre at sann gjennomsnittlig ratio med høy sannsynlighet ligger innenfor området 0.80-1.25. Den faste definisjonen er altså 0.80test/AUCreferanse <1.25
og tilsvarende for Cmax. En grense på ±20 % anses normalt som akseptabel når man ser den i sammenheng med variasjonen i absorpsjon, distribusjon, metabolisme og eliminasjon mellom individer og for hvert enkeltindivid fra dag til dag. Retningslinjer for bioekvivalensstudier (2) åpner for at et videre område (±25 %) kan godtas for enkelte produkter, men dette må begrunnes. For legemidler med et smalt terapeutisk vindu kan et smalere ekvivalensområde kreves (±10 %).
Figur 1 viser eksempler på 90 % konfidensintervall for gjennomsnittlig ratio mellom test- og referanseprodukt i sju forskjellige situasjoner. I eksempel 1-3 og 7 er testproduktet ikke bioekvivalent med referanseproduktet fordi minst en av grensene i 90 % konfidensintervallet ligger utenfor det godkjente området. I eksempel 4-6 er produktene bioekvivalente fordi hele intervallet ligger innenfor 0.80-1.25.

Figur 1. Eksempler på resultater av sju forskjellige bioekvivalensstudier. Fylte sirkler angir estimert gjennomsnittsratio for hver studie og blå linjer lengden av hvert 90 % konfidensintervall. Nummer 1-3 og 7 viser situasjoner hvor testprodukt ikke anses som bioekvivalent med referansen fordi minst én av grensene i 90 % konfidensintervallet ligger utenfor det godkjente området. I eksempel 4-6 er produktene bioekvivalente fordi hele intervallet ligger innenfor området 0.80-1.25.
Forsøksplan
Bioekvivalensstudier utføres vanligvis med friske, frivillige forsøkspersoner. Det er vanlig å inkludere n≥12 personer, og det er vanligvis kun for legemidler med høy variasjon i farmakokinetikk at det inkluderes mer enn n=30. Forsøkene er alltid overkrysningsforsøk; det vil si at alle forsøkspersonene får både referanseprodukt og testprodukt. Hver forsøksperson sammenliknes altså med seg selv, og når vi skal trekke slutninger blir den intraindividuelle variasjonen viktigere enn variasjonen mellom individer. Oftest består forsøket av to perioder, og forsøkspersonene får produktene i tilfeldig rekkefølge. Sjelden utføres det forsøk med tre eller fire perioder slik at den intraindividuelle variansen for henholdsvis test- og referanseprodukt kan estimeres. Normalt anses det tilstrekkelig å utføre enkeltdosestudier, men hvis det aktuelle virkestoffet har en dose- eller tidsavhengig farmakokinetikk, kan forskjellen mellom preparater være større ved steady-state, og studier med multiple doser kan kreves.
Statistisk analyse
De enkelte konsentrasjonsmålingene inngår ikke direkte i den statistiske analysen. Hver plasmakonsentrasjonsprofil oppsummeres ved AUC og Cmax (og eventuelt Tmax). Variablene AUC og Cmax logaritmetransformeres før data analyseres. Dette vil typisk gjøre variasjonen tilnærmet normalfordelt, og såkalte parametriske metoder, for eksempel variansanalyse (ANOVA) kan benyttes. Ved bruk av en slik modell tar man hensyn til både inter- og intraindividuell variasjon. Dessuten sammenliknes ikke bare AUC eller Cmax med hver av de to behandlingene direkte; man justerer også for periode- og sekvenseffekter. En periodeeffekt innebærer at det kan være forskjell mellom de to tidspunktene legemidlene er testet på, og en sekvenseffekt innebærer at det er forskjell mellom personer som får referanseprodukt først og deretter testprodukt og personer som får produktene i omvendt rekkefølge. Man vil ikke forvente slike effekter dersom forsøket er godt gjennomført. Dersom man finner slike, bør man lete etter en god forklaring.
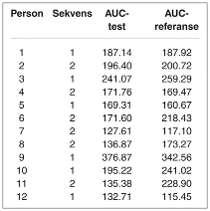
Tabell 1. Eksempel på AUC-verdier for 12 forsøkspersoner. Sekvens angir rekkefølgen produktene er gitt i. 1= test først, 2= referanse først.
Et eksempel
Tabell 1 gir et eksempel på resultater fra en bioekvivalensstudie. Som vi ser av tallene, avviker AUC betraktelig for testprodukt sammenliknet med referanse for noen forsøkspersoner (for eksempel nummer 6, 8 og 11). Variasjonen mellom individer er også stor. Dette er vanlig i bioekvivalensstudier.
Som beskrevet tidligere er det vi trekker slutninger om i en bioekvivalensstudie den gjennomsnittlige ratioen AUCtest/AUCreferanse, og som nevnt beregner vi gjennomsnittsratioen via logaritmetransformasjon (geometrisk gjennomsnitt). I eksempelet blir estimert gjennomsnittsratio 0.926. Det ser altså ut til at AUC i gjennomsnitt er litt lavere for det nye produktet enn forreferansen. Vår beste «gjetning» er at gjennomsnittlig AUC for testproduktet er 92.6 % av AUC for referansen.
Vi kan likevel ikke trekke en slutning kun på grunnlag av dette estimatet. Et 90 % konfidensintervall for sant geometrisk gjennomsnitt må også beregnes. I eksempelet blir dette 0.841-1.020. Fordi dette konfidensintervallet ligger innenfor grensene 0.80-1.25, kan de to produktene anses som bioekvivalente.
Fortolkning
For å fortolke bioekvivalens må man forstå hvilken informasjon som egentlig ligger i et 90 % konfidensintervall. Definisjonen av et konfidensintervall er «et område som med høy sannsynlighet inneholder det sanne gjennomsnittet». Når vi beregner et gjennomsnitt, er dette kun å betrakte som et anslag for sann verdi i den populasjonen vi ønsker å trekke slutninger om. Hvis vi hadde gjort den samme studien om igjen, ville estimert gjennomsnitt ikke ha blitt nøyaktig det samme som første gang, men trolig noe i nærheten. I teorien kan vi tenke oss at vi utfører en studie mange ganger og beregner et stort antall gjennomsnitt. De fleste av disse vil ligge i nærheten av sannheten, mens noen få vil ligge langt unna. Problemet i praksis er at vi bare estimerer ett gjennomsnitt, og vi vil ikke vite om dette er nær sannheten eller ikke.
Et konfidensintervall benyttes til å måle usikkerhet. Før vi beregner konfidensintervallet vet vi at et 90 % konfidensintervall med 90 % sannsynlighet vil inneholde den sanne verdien av gjennomsnittet. Etter at det er beregnet, gir det ikke lenger mening å si at det er 90 % sannsynlig at det inneholder sannheten - enten er sann verdi innenfor grensene eller den er det ikke. Ofte sier man at et 90 % konfidensintervall inneholder plausible verdier for hva som kan være sant gjennomsnitt.
Overfører vi dette til bioekvivalenseksempelet, ser vi at med et 90 % konfidensintervall som er estimert til 0.841-1.020, er det dokumentert at det sanne gjennomsnittet med høy sannsynlighet ligger innenfor grensene 0.80-1.25. Bioekvivalens er dermed demonstrert. Dette betyr ikke at ratioen mellom de to produktene ligger mellom 0.80 og 1.25 for alle enkeltindivider. Som for kliniske studier av effekt og sikkerhet trekker vi slutninger om gjennomsnittet i en populasjon, ikke om effekten for en enkelt forsøksperson eller pasient.
En bioekvivalensstudie tester altså det vi kan kalle forskrivbarhet. Dersom vi er interessert i å teste om legemidler er byttbare på individnivå, ville individuell bioekvivalens vært av betydning. Individuell bioekvivalens estimeres normalt ikke i bioekvivalensstudier som danner grunnlag for en søknad om markedsføringstillatelse.
Referanser
- Nitteberg-Sørensen, B. og Madsen S. Bioekvivalens og medisinsk likeverdighet. Norsk Farmaceutisk Tidsskrift, nr 2, 17-18, 2009.
- CHMP guideline on the investigation of bioequivalence.
www.emea.europa.eu/pdfs/human/qwp/140198enrev1.pdf
(Publisert i NFT nr. 4/2009 side 18–19.)